




From EU medical device regulation (MDR) to NMPA (chinese authority)

Fondo
In this article we want to reveal the spirit to register medical device in China from MDR (EU manufacturer).
The key to be successful at international registration is to know the country specific requirements and regional registration pathways.
It is first to know that most Chinese registration is based on home country approval. Manufacturer doesn’t start Chinese registration from zero, but can profit from a broad MDR (or former MDD) complex. The design history file from R&D and device master record is nearly unchanged as a basis. Depending on the product group, you could have a Chinese variant of medical device with Chinese labelling, local testing (in China) with Chinese standard and oft with Chinese clinical evaluation. The trouble and pain is how to split an EU technical document to fit structured electronic folder of Chinese technical document.
Vía de registro chino
1. Legislativo
European Union with 27 member states issues the law Medical Device Regulation (MDR) starting in 2017. However the individual notified bodies are assigned for product and system certification. National Medical Products Administration (NMPA) issues the regulation of medical device in China and its sub-organisation reviews and approves registration dossier.
La Ordenanza 739 del Consejo de Estado, con 107 artículos, emitida en 2021 por la autoridad china NMPA, es equivalente al Reglamento Europeo de Dispositivos Médicos (EU MDR), que consta de 123 artículos y 17 anexos. En China existen normativas complementarias que regulan el registro, las Buenas Prácticas de Fabricación (BPF), la clasificación, la evaluación clínica, la vigilancia posterior a la comercialización y la documentación técnica, entre otros aspectos. Además, existen normas y guías generales y específicas para cada producto.
2. Chinese agent (& EU authorised representative and Person Responsible for Regulatory Compliance)
Chinese agent is as liaison between foreign manufacturer and Chinese authority. Chinese agent has more responsibility than authorised representative in EU however less liability for product in the market. There is no role as Person Responsible for Regulatory Compliance at manufacturer required in China.
Besides the following responsibilities, Chinese agent has an most important role to submit the technical document to authority and to obtain product approval.
• Communication with NMPA
• Report of adverse event
• Assistance of the potential inspection by authority
• Transfer of regulatory update

3. Clasificación y código de producto
In China there is a different classification system more similar to FDA than to EU. The same is the risk-based classification of medical device. Depending on intended use you can match to right 6-digits product code which automatically has its classification (I, II and II). The higher the classification is, with more demanding is technical document to submit.
4. Certificación del producto únicamente
In EU there are 3 years certification cycles for quality management system, very often in combination of MDR and ISO 13485. To register product in China, manufacturer only has to submit valid ISO 13485 certificate. In theory NMPA may make a remote or onsite overseas inspection of foreign manufacturer which is rare.
So Chinese registration is a pure product certification. Unlike in EU (MDCG 2019-13 Guidance on sampling of MDR Class IIa / Class IIb and IVDR Class B / Class C device), all manufacturers of class II and III medical device has to submit each technical document in China. For class I medical device there is only an easy filling system.
En China existen principalmente tres tipos de registro: inicial, de modificación y de renovación. Dado que el certificado chino tiene una validez de solo 5 años, el fabricante debe renovarlo cada 5 años si no hay cambios significativos (similar al artículo 120 del Reglamento de Dispositivos Médicos de la UE).
“Risk” is frequent mentioned at MDR. In China although it is rarely written at highest law of medical device, still there is a risk-based registration benefiting from clear product code for general group. Only some of high risk products need clinical trial or complex clinical evaluation.
5. Documento técnico
In accordance to guidance “Non-In vitro Diagnostic Device Market Authorization Table of Contents” at International Medical Device Regulators Forum (IMDRF), NMPA published in 2021 guidelines for electronic submissions with requirements aligning with international table of content (toc) for all submission format. The submission dossier in China is in structured electronic toc and in Chinese language. Derived from Annex II at MDR manufacturer has a good reference dossier to transfer to Chinese TD. However often due to Chinese requirements manufactures have to re-puzzle the contents in Chinese dossier. There are 6 main chapters (CH x) of Chinese TD.

6. Informe de pruebas locales y evaluación clínica
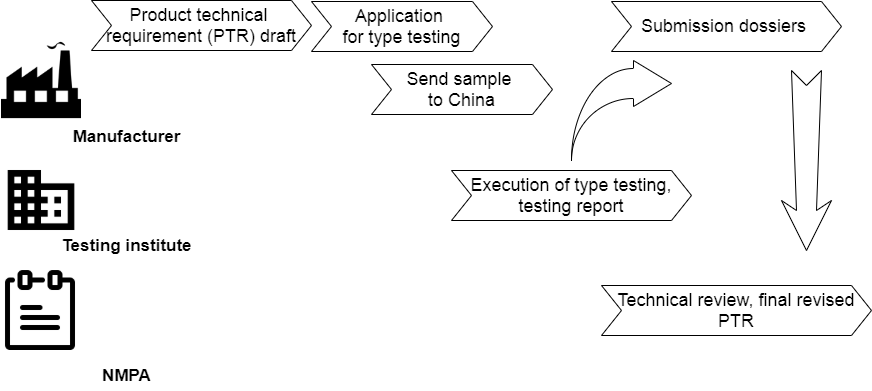
Special at Chinese registration is local testing. It is also called in-country testing or product/sample testing.
Además de preparar los expedientes de presentación en papel para el registro del producto, el dispositivo médico extranjero debe enviarse a institutos de pruebas acreditados por la NMPA en China después de redactar los "requisitos técnicos del producto" (PTR).
El informe de evaluación clínica (IEC) en China presenta tres escenarios: IEC simplificado en tabla, IEC basado en dispositivos equivalentes o similares y ensayos clínicos chinos. En el segundo escenario, los fabricantes deben comparar dispositivos médicos equivalentes aprobados en China y consultar la literatura china. En el tercer escenario, se observa una tendencia positiva hacia la aceptación total o parcial de estudios clínicos realizados en el extranjero.
7. Presentación y revisión de documentos técnicos
The review process of medical device in EU is perhaps a sensible topic. Each NB has its own charisma.
La autoridad competente, dependiente de la NMPA, es la encargada de la evaluación de dispositivos médicos chinos (CMDE, por sus siglas en inglés), que certifica la documentación técnica electrónica de los productos importados de clase II y III fabricados fuera de China.
In China the review time by authority is 153 working days for class 2 MD and 183 working days for class 3 MD (*20 working days are one month). The authority set every year new records to reduce review days than official needed.

8. Aprobación y vigilancia posterior a la comercialización
Tras la aprobación del producto, el fabricante deberá finalizar el etiquetado en chino con el número de certificación. Al igual que con Eudamed en la UE, el fabricante deberá cargar el UDI y el código de emisión en una base de datos china diferente.
The post market surveillance is less complex in China than in EU (see below). For sure the workflow and deadline to report vigilance is in China way different and in rapid march in number.

Zero to positive impact of MDR on Chinese registration
Etiquetado de la UE
Compared MDR annex I, chapter III (Labelling) to Chinese guidance of labelling, there are only nice to have contents on EU labelling. Manufacturers can keep a variant of Chinese labelling fitting Chinese requirement or transfer new MDR contents one to one to Chinese labelling.
Post market surveillance
This is the most challenging part at MDR underscoring patient safety. The market surveillance report in EU acts a good basis to provide new annual quality management report and periodic risk evaluation report in China. If overseas clinical trial was existing, it could assist the potential requirements of clinical trial in China. The degree of difficulty of Chinese Post market surveillance is developing and raising.
Capacity of authority
A diferencia del organismo notificado de la UE, la autoridad china es gubernamental, emite regulaciones y verifica todos los dispositivos médicos importados. Cuando se publica una nueva regulación, la autoridad no tiene excusa para la transición ni capacidad de respuesta. Al igual que la FDA, la autoridad china se esfuerza por batir récords en el tiempo de revisión de expedientes.
Reclassification
There is a trend at MDR that some products are up-classified. Actually it has no impact on Chinese classification because in China there is own classification rule and product code system.
Change assessment
La guía para cambios significativos es la MDCG 2020-3, Guía sobre cambios significativos en relación con la disposición transitoria del artículo 120. Se centra en los productos que pasan de la Directiva de Dispositivos Médicos (MDD) al Reglamento de Dispositivos Médicos (MDR). Dependiendo del organismo notificado, la notificación y revisión de cambios significativos en la UE varía. Rara vez es necesario volver a presentar la documentación técnica completa tras un cambio significativo.
There is only system of new change registration upon significant change of products or record minor change in QM system in China. No context in term of product change is between 2 markets.
Impacto de la transición MDD-MDR
Declaration of conformity
CE certificate is typical recognised as home country evidence for EU manufacturer in China. Independent of classification and transition of MDR, at time of submission to Chinese authority, the declaration of conformity at EU manufacturer should be valid.
EU 2023/607
So far there is no notification from Chinese authority whether they accept the agreement of notified body with manufacturer to accept MDD certificate valid upon any conditions. However it is expected that the transition of MDD products will be accepted in China.
In the case of MDD certificate with inactive (obsolete product) which is valid till e.g. Dec, 2018, manufacturer can outsource production in China. There are 2 options:
1. For in China approved product with no MDR future, manufacturer could make an accelerated “local to transfer” registration and have an own entity to produce medical device after approval (additional Chinese GMP needed).
2. For never in China approved product with no MDR future, manufacturer could start a new registration and outsource third party in China to produce medical device quasi outsourcing Chinese production.
For both options at the end of approval, the product is made in China and could profit a lot as local product at tender.
Perspectiva
Either you have technical document compliant to MDD or MDR, contact us e.g. in term of on-demand workshop to have in-depth gap analysis.
O si desea obtener una sólida formación en registro chino, visite nuestra universidad en línea de 1 año con matrícula gratuita.
