




China competes against US in medtech (NMPA & FDA)

Fondo
This article is written for all personal interested and working on 2 major medical device markets in the world China and US. Unlike politic conflict, there are many friendly similarities in these 2 most complex agencies from legislative to registration.
We want to compare US and Chinese markets of medical device in an easy and competitive way. From our view, Chinese authority National Medical Products Administration (NMPA) has followed a lot from the most modern U.S. Food and Drug Administration (FDA) and expanded and improved even a bit with Chinese elements.
Actually NMPA had a former American name Chinese Food and Drug Administration back in 2014 when the basic legislative is in form initialled. Since 10 years the regulations are developing in a crazy rapid tempo and high-quality regulating tone.
Compared to NMPA, FDA has its Federal Food, Drug, and Cosmetic Act (FD&C Act) already in 1938. Its legislative and standards are still state of the regulatory art in global compliance.
Registration scores (USA & China)
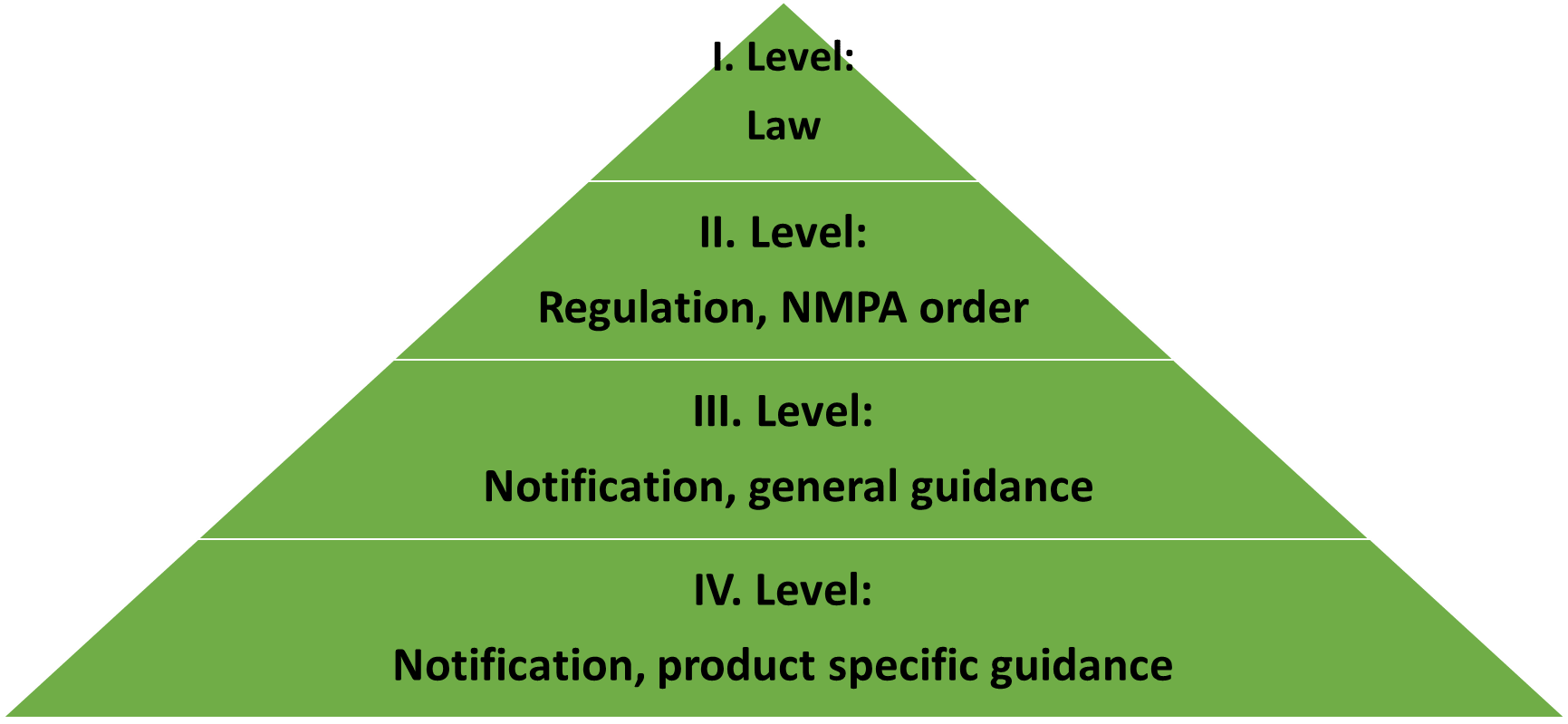
1. Legislative, 1:2
En Estados Unidos, la legislación sobre dispositivos médicos se encuentra en el Título 21, Capítulo 9 y Subcapítulo V: Medicamentos y Dispositivos de la Ley Federal de Alimentos, Medicamentos y Cosméticos (FD&C Act) del Congreso de los Estados Unidos. El Código de Regulaciones Federales (CFR) es publicado por la agencia reguladora federal correspondiente (FDA). La referencia a los dispositivos médicos se encuentra en el Título 21, Partes 1-99 y 800-1299. Existen dos recursos para consultar la legislación sobre dispositivos médicos, que a menudo se mezcla con la legislación sobre medicamentos en Estados Unidos.
The only binding law of medical device in China is newest regulations on Supervisory Management of Medical Devices in 2021. Since then diverse provisions, notifications and guidances are published which should be comply with and followed in the whole life cycle of medical device marked in China.
Existen directrices y normas de apoyo que estipulan los requisitos de los dispositivos médicos en ambos mercados y que desempeñan un papel fundamental en el desarrollo y el registro de productos.
Due to Chinese language and regulation sometime brief formulated and in lower edition it makes difficult to understand Chinese legislative. In term of difficulties of legislative China wins.
2. Sistema de gestión de la calidad, 2:1
En EE. UU., el sistema de calidad se rige por la norma QMSR, que actualmente se está alineando con la norma ISO 13485.
En China, para los productos fabricados en el país, el fabricante debe solicitar la certificación de Buenas Prácticas de Fabricación (BPF) chinas, conforme a las "Medidas para la Supervisión y Administración de la Producción de Dispositivos Médicos". Para los productos fabricados en el extranjero, se requiere una certificación internacional del sistema de gestión de calidad únicamente para el registro del producto, como la ISO 13485 o el Informe de Sistemas de Gestión de Calidad (QMSR) de la FDA.
From the view of foreign manufacturers, entering US market means to align additionally to QMSR with a few efforts. US beats China with more QM effort.
3. CN & US agent, 1:1
For foreign manufacturers in both markets, you need a legal person there. In US it is called US agent, in China of course Chinese agent. They are quasi a regulatory representative for foreign manufacturer. From our practise, both agents are essential to submit electronic technical documentation and to communicate with authority by dossier deficiency.
In the reality after product approval, Chinese agent has more actions to coordinate as vigilance, to register of UDI and issuance code. Normally foreign manufacturer completes after approval actions on its own in US, maximal with help of US correspond.
4. Product code and classification, 1:1
En EE. UU. existen 16 paneles de clasificación con números de regulación de 7 dígitos que abarcan 1700 tipos de productos. Paralelamente, existe un código de producto de 3 dígitos.
China ha transformado el código de producto estadounidense a un código de 6 dígitos. Detrás de estos 6 dígitos se ocultan tres niveles de categoría. En total, existen 22 grupos principales de productos.
Knowing regulation number and product code in US you can find classification, submission type, standard, GMP requirements.
Knowing product code in China you can match classification, guidance, Chinese standards and clinical pathway.
En ambos mercados existen 3 clases de riesgo (I, II y III), que se determinan según el uso previsto del dispositivo médico.
Cuanto mayor sea la clasificación del producto, más compleja será la documentación técnica a presentar. Para los dispositivos médicos de clase I, en ambos mercados solo existe un sistema de presentación sin proceso de revisión.

5. Documento técnico, 1: 1
Ahora nos encontramos en la etapa de presentación de la documentación técnica electrónica. Cada mercado tiene su propio sistema electrónico y estructura de carpetas para archivar la documentación técnica. Queremos limitar la comparación entre los dispositivos médicos típicos de clase II/III en China y la notificación previa a la comercialización (510k) en Estados Unidos.
Surprisingly under main sections or chapters between US and Chinese market there are huge similarities.
Para la certificación US 510(k), se declara como equivalente sustancial a otro dispositivo comercializado legalmente en EE. UU. Esta filosofía de equivalencia se encuentra en la comparación con el dispositivo de referencia y la evaluación clínica en China.
Los requisitos de documentación técnica en ambos mercados son transparentes y predecibles.

To master product registration, it is the key to know the heavy weights to be examined by authority. We explain below product testing and clinical evaluation.
6. Pruebas de registro, 1:2
Las pruebas regionales consumen mucho tiempo y dinero. En EE. UU., hay que enviar el dispositivo para que un ciudadano estadounidense lo someta a una validación de usabilidad. Para los productos inalámbricos, no hay que olvidar las pruebas con la Comisión Federal de Comunicaciones (FCC).
In China for class II and III medical device, manufacturer has to send device to China by from state accredited testing institutes. No testing unit should fail with operation of Chinese engineers at testing institutes. The performance and functional specifications should not deviate than required in national and industrial standards or by manufacturer own stated.
Resulta complicado enviar dispositivos médicos de fabricantes extranjeros a China para que se sometan a pruebas conforme a las normas chinas, que no siempre coinciden con las normas internacionales más recientes. En lo que respecta a las pruebas de registro de productos, China presenta mayores dificultades que Estados Unidos.
7. Evaluación clínica, 0:1
In US there is no clinical evaluation. For some high risk devices, clinical study is needed.
En China, los requisitos para la evaluación clínica son similares, pero menos estrictos que los de Europa (MDCG 2020-13). Para la evaluación clínica, el fabricante debe comparar su dispositivo con uno propio o de un tercero aprobado en China. Además, solo para algunos dispositivos de alto riesgo, el fabricante debe realizar un estudio clínico en China. Con frecuencia, los datos clínicos obtenidos en el extranjero se reconocen en China.
Dado que en China se debe realizar una evaluación clínica especial que cumpla con los requisitos chinos, la exigencia clínica es mayor en China que en Estados Unidos.
8. Tiempo de revisión y respuesta del TD, 1: 1
We compared the review time by authority of typical registration at FDA 510k with class II and III medical device in China.
Como se observa en la figura, la FDA debería completar su revisión en un plazo de 100 días hábiles para la solicitud 510(k) y en un plazo de 180 días hábiles para la aprobación previa a la comercialización (PMA). En China, el tiempo de revisión para dispositivos médicos de clase III es prácticamente el mismo que para la PMA en Estados Unidos. La autoridad china requiere más tiempo (155 días hábiles) para dispositivos médicos de clase II que para la solicitud 510(k).
En cuanto al tiempo de respuesta en caso de deficiencia por parte del fabricante, la autoridad china permite un plazo menos estricto de un año para responder, en contraste con los 180 días hábiles que se requieren para obtener información adicional en los EE. UU.


9. Official Cost, 1:1
Comparamos el coste oficial de los registros típicos en China y en Estados Unidos.
En EE. UU., para las pequeñas empresas el costo es económico. Además del registro de establecimiento, el fabricante debe pagar anualmente, en 2024 $7,653.
In China we list price of typical registrations below. Change registration is as new 510k upon substantial change. Because the Chinese certificate is only 5 years valid, it must be renewed every 5 years.
| Clase de producto | Tipo | Costo oficial (dólares) |
|---|---|---|
| Clase I | Registro inicial y de cambios | Gratis |
| Clase II | 29.700 | |
| Modificar el registro | ||
| Modificar el registro | ||
| Registro de prórroga (cada 5 años) | 5.750 | |
| Clase III | ||
| Modificar el registro | ||
| Registro de prórroga (cada 5 años) | 5.750 | |
| Ensayo clínico | 6.090 |
10. PMS, 1:0
In term of overseas inspection so far there are more possibilities to be inspected by US authority than Chinese authority on site of manufacturers. However after medical device is placed in China, in all supply chains products can be chosen to test upon requirements already proved at registration.
En términos de vigilancia, existen diferentes plazos y procesos en ambos mercados, por lo que resulta injusto distinguirlos.
In term post market surveillance, in US is more passive in product risk-based approach. FDA can order two primary types of mandatory postmarket studies: so-called “522 studies” (also referred to as postmarket surveillance studies) and PostApproval Studies (PAS).
En China, el fabricante debe presentar activamente un informe anual de autoevaluación del sistema de calidad y un PSUR chino, que forman parte de las actividades posteriores a la aprobación.
Summarised after product approval, manufacturer has to maintain post market more intensively for potential FDA than NMPA. In China there are increasing post market monitoring. It is still not that difficult to get brief report approved.
Conclusion
The final score is US & China 10: 11. We highly recommend manufacturer to welcome these attractive markets. Through a demanding registration is best way not only for future sales making innovative products quicker to patient but also a chance to prove safety and effectiveness to generate next product generation.
Do you want to know more insight more than these main registration factors, you can visit our one year academy with personal coach.
¿Qué tal si nos elige como gestores interinos? Nos apasiona comprender el panorama global del cumplimiento normativo. Queremos batir el próximo récord de registro junto a ustedes.
