



美国FDA注册
关键词
美国食品药品监督管理局,医疗器械,Federal Food, Drug and Cosmetic Ac,联邦规则汇编,指导原则,上市前通告 510k, 市前批准PMA , 提交前咨询, Additional Information Request,唯一器械标识,美国代理人,可用性检验,nationally recognised testing laboratory

美国医疗器械法规入门
最高权威机构
美国食品药品监督管理局(Food and Drug Administration,FDA)的主要职能为负责对食品、膳食补充剂、药品、疫苗、生物医药制剂、血液制剂、医疗设备、放射性设备、兽药和化妆品进行监督管理。他们权利要远大于中国的国家药品监督管理局。其中相当于医疗器械技术审评中心的美国审评机构是设备仪器与放射健康中心 (Center for Devices and Radiological Health,CDRH)。
医疗器械法规,指导原则,标准
美国国会在1938年通过联邦食品、药品和化妆品法案 (Federal Food, Drug, and Cosmetic Act)。其中标题21第九章第五分章关于医疗器械和药物。建议大家根据关键词个人化地搜索,保存,使用这个历史最悠久的医疗器械法规。另一个查阅医疗器械法规的是联邦规则汇编(Code of Federal Regulations)。其中21 CFR Parts 1-99和21 CFR 800-1299是FDA级别关于医疗器械的行政法规。大家最熟悉的莫过于质量管理体系21 CFR 820.
坦白说中国的指导原则和标准的精髓来源于美国。根据不同产品类型的注册经验,药监局发布了很多针对产品类型或通用产品的指导原则和标准,有利于制造商在准备申请材料时有法可依。美国更简单的是,如果您知道医疗器械的三位编码,就可以在法规库找到所有适合的指导原则和标准。FDA礼貌地称指导原则和标准为推荐性,虽然没有强制性的属性,但制造商最好按照指导原则和标准做全应有的产品检验.
质量管理21 CFR 820
21 CFR 820是FDA关于质量体系的法规,医疗器械注册后制造商有被FDA检查生产现场的可能性,非美国的制造商根据产品不同被查的几率相对不太高。如果查厂出现问题,会发补警告信warning letter.
21 CFR 820的内容和各国的质量体系法规甚至跟中国的GMP非常相近,只是根据审查机构的不同,侧重点不同。目前FDA在转变成国际通用的ISO 13485,取代21 CFR 820。
质量管理21 CFR 820
§ 820.20 - 管理责任
§ 820.22 - 质量审计
§ 820.25 - 人员
§ 820.30 - 设计控制
§ 820.40 - 文件控制
§ 820.50 - 采购控制
§ 820.60 - 身份证明
§ 820.65 - 可追溯性
§ 820.70 - 生产和过程控制
§ 820.72 - 检查、测量和测试设备
§ 820.75 - 过程验证
§ 820.80 - 接收、过程中和成品设备验收
§ 820.86 - 验收状态
§ 820.90 - 不合格产品
§ 820.100 - 纠正和预防措施
§ 820.120 - 设备标签
§ 820.130 - 设备包装
§ 820.140 - 处理
§ 820.150 - 存储
§ 820.160 - 分发
§ 820.170 - 安装
§ 820.181 - 设备主记录
§ 820.184 - 设备历史记录
§ 820.186 - 质量体系记录
§ 820.198 - 投诉文件
§ 820.200 - 服务
§ 820.250 - 统计技术
注册类型
美国有以下类型的注册。
- 研究器械豁免 (IDE)
- 上市前通告 (Premarket Notification,510(k))
- 上市前批准(Premarket Approval,PMA)
- De Novo
- 人道主义器械豁免 (HDE)
最重要的是大多二类医疗器械的上市前通告510k和大多三类医疗器械的上市前批准PMA。如果二类产品没有任何风险,还可以免上市前通告510k exept,注册难度接近一类产品。一类医疗器械,制造商只需要登记,不需要任何申请材料和审批。De Novo类似中国的创新产品的绿色通道申请。研究器械豁免 (IDE) 允许研究器械用于临床研究,以收集安全性和有效性数据。只有通过了临床试验才允许申请510k或PMA。
以下是官方关于510k的授课视频

特殊管理局的咨询
美国FDA最专业的之一是一流的咨询服务质量。这种咨询曾为Q-Submission。
其中最常见的是
1.Pre-submission,可以当面咨询也可以电话式咨询。Pre-submission一般在准备注册材料是,制造商询问FDA专家的一些疑难。
2. 还有一种咨询是Submission Issue Requests (SIRs),在注册提交后针对在发补过程中的问题解答,针对以发补的通知为基础。
3.Study Risk Determinations是对计划的临床试验的风险进行交流。
如果您想得到专业的FDA回答,也应该利用这次与专家交流的机会认真准备问题,让FDA理解您的产品懂得您的问题。通常需要递交的材料是
· 目录
· 咨询的概括
· 首选的反馈形式
· 医疗器械的完整描述,建议的预期用途
· 计划的检验策略
注册费用和审评时间
以下是2023年几种典型注册的费用。其中可以看出微型企业,也就是最近一个纳税年度的总收入和销售额低于 1 亿美元的企业,可以享受大打折扣的注册费用。
申请类型 标准费用 微型企业的费用
510(k) $19,870 $4,967
513(g) $5,961 $2,980
PMA $441,547 $110,387
我们简单介绍典型510k的审评时间。递交产品技术文件后,最重要的一环是FDA的发补。制造商应该在技术文件提交后第7天交付注册费用,FDA会在第15天确定技术文件的完整性。45天后,第60天FDA完成技术审核,发补问题Additional Information (AI) Request。制造商必须在180天内给予回答。此时FDA审核时间暂停。等到发补回答呈交后审核时钟继续,通常第90天(复杂申请100天)为里程碑, FDA会宣布注册是否被批准。美国医疗器械没有注册证,FDA会给予与已上市产品实质上相同决定电子信件。
传统510k的审评时间

美国注册的流程
技术文件差异可行性分析
根据您的医疗器械的特点,预期用途,已有的技术文件,我们会进行专业的差异性分析。如果已有中国或欧洲的成功注册经验,其实最重要的是补充差异的检验及英文文件。美国510k的特色是有已上市的同类产品进行比较,所以关键是考虑510k注册的可行性。如果问题比较繁琐,建议参加FDA的免费预咨询,把注册的难点提前做好分析。
选择美国代理人
像中国代理人一样,美国代理人是申请美国注册的前提,负责FDA与美国以外制造商的交流。美国代理人得居住在美国或在美国设有营业场所。代理人的其它责任是协助可能的国外查厂。与中国代理人不同,代理人没有责任参与市场后不良事件的报告,也不需要为制造商登记任何产品,唯一器械标识信息。最奇绕的是美国代理人没有义务递交申请材料,意味着制造商完全可以利用自己的能力或另外咨询公司完成电子注册材料的申请,美国代理人只不过作为第一联系人。
所以最好在注册前期选择好美国代理人。
确定产品分类,注册类型
美国有着类似中国的产品号码,由三位字母组成。通常起决定产品号码的是医疗器械的预期用途。
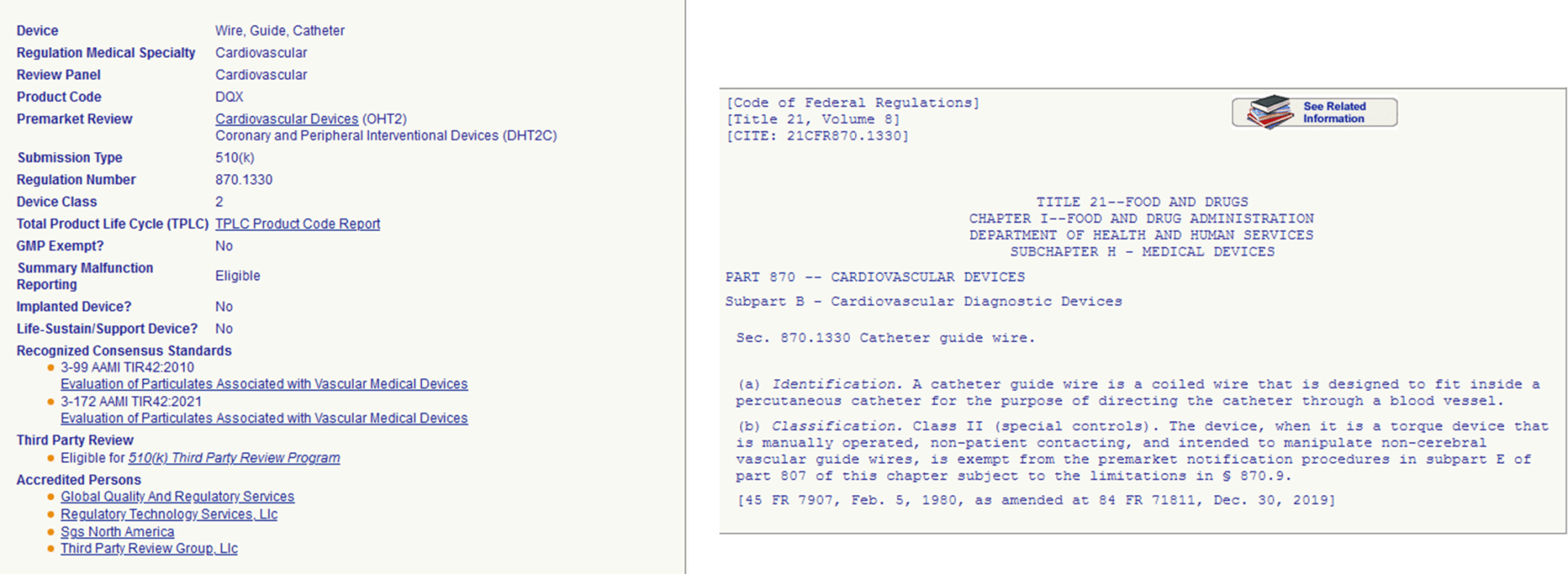
以下是举一个最常见的的二类导管例子,产品号码DQX,法规号码870.1330. 注册类型是510k,以美国产品号码搜索还会查询到产品需要遵守的标准。通过法规号码870.1330,可以得到联邦规则汇编(Code of Federal Regulations)在这个法规号码要求的医疗器械范围。
导管的产品号码,法规号码 和注册类型是
产品检验
美国没有中国式的产品检验,必须通过国家认证的检验机构。但是制造商面临着在中国的检验报告是否会被FDA承认。我们建议制造商,最基本的安检和电磁兼容性检验应该获得有资质的检验所的英文报告,检查报告里面引入的是国际或美国的医疗器械标准。希望制造商考虑关键的检验在美国进行。
1. 可用性检验针对绝大数医疗器械不可或缺,法规是Applying Human Factors and Usability Engineering to Medical Devices。可用性检验通过用户的使用来评估产品是否满足用户需求的技术。由于要求是美国公民作为产品使用者进行测试,最保险的是把医疗器械寄到美国进行测试。产品检验发生的使用错误与风险报告密切相关。法规里有可用性检验报告的框架,值得在测试前后参考。
2. 另外很多医院只接受含有NRTL标识的医疗器械,这意味着医疗器械得通过所谓国家承认的检测所(nationally recognised testing laboratory, NRTL)的安检和电磁兼容性检验。通过NRTL检测后,制造商还会面临着规律的年审,以持续有效的证书。
3. 联邦通讯委员会Federal Communications Commission, FCC只针对射频无线医疗设备,例如Wi-Fi、蓝牙、RFID。虽然对医疗器械审批监督的是FDA,但是要进入美国市场,另一个机构签发的FCC的认可有备无患。
通过NRTL, FCC,制造商有义务添加代表获得证书的标识

完整技术文件, 电子注册
以下是传统510k的章节。可以看出,绝大数章节都是和中国注册材料极为相似。最终的技术文件从2023年10月都已电子文档的形式递交,称为电子提交模板和资源 (electronic Submission Template And Resource, eSTAR)。

登记医疗器械制造商和市场后监督
注册通过后,制造商应登记其实体,医疗器械,唯一器械标识,完善质量体系21 CFR 820,根据21 CFR 801确保合规的标签,说明书甚至网页关于特殊注册美国产品的内容.
医疗器械的市场后监督主要是不良事件和召回21 CFR 803 and 806,没有任何按期主动上交的市场后评价文件。
例外的是被通知上交的 522上市后监督研究,针对的器械:
· 失败很可能会对健康造成严重的不良后果
· 在儿科人群中发挥重要作用
· 用于植入体内一年以上
· 在器械用户设施外使用的生命维持或生命支持
注册美国的优势
很多国家跟美国FDA有着协议承认美国的注册,或者像瑞士,直接承认美国注册,不需任何另外的注册审批过程。或者像墨西哥, 新加坡, 韩国等,医疗器械如果美国市场已经注册,当地药监局会缩短审评时间。即使不递交注册美国的证明,很多市场的注册文件包括已注册市场清单。主管部门会清楚,已在美国注册的医疗器械不仅通过严谨的审评,而且技术文件包括非常专业的产品验证,审批部门会照顾这个有经历的热门种子号产品。
像上面分析的技术文件,通过美国注册,相当一部分的高质量以及符合最先进标准的检验可以无后顾之忧地用于几乎任何有难度的器械市场的申请。
值得追踪的法规链接
Law
CFR 820
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=820
510k database
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm
PMA
https://www.fda.gov/medical-devices/premarket-approval-pma/pma-review-process
Standard
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/search.cfm
Testing
https://www.fda.gov/medical-devices/digital-health-center-excellence/wireless-medical-devices
Cost and time
https://www.fda.gov/industry/fda-user-fee-programs/medical-device-user-fee-amendments-mdufa
Change
Learn (Video)
https://www.fda.gov/training-and-continuing-education/cdrh-learn
为什么选择我们的服务
我们有着国际注册的经验,在熟悉您产品技术文件的基础上,更容易进行申请材料的控制,对难度大的检验多加照顾。
由于我们的梯队也在美国,您可以信任让我们作为代理人,在注册任何阶段与FDA咨询,在进入市场后跟您的经销商处理不良事件等。
更重要的是我们熟悉中国的法规背景,更容易对产品文件和质量体系进行美国转化。我们的双语式项目管理,让您从始至终洞察着注册进展,并有效地完成高质量的发补回答,在承诺的时间完成注册。